
식품의약품안전처가 의약품 산업 경쟁력 강화에 나선다.
식약처는 국제적으로 표준화 추세인 ‘의약품 설계기반 품질(QbD)’ 제도를 의약품 허가심사 평가 제도에 도입하는 등 ‘의약품의 품목허가ㆍ신고ㆍ심사 규정’을 30일자로 개정ㆍ시행한다.
이번 개정의 주요 내용은 △의약품 설계기반 품질(QbD) 적용 품목의 변경허가 간소화 △임상ㆍ비임상시험 자료 표준형식(CDISC) 제출 근거 마련 △신약 품목허가 상담 신청제도 도입 △수입 신약 제출자료 간소화 등이다.
우선, QbD 적용 품목의 변경허가를 간소화한다. 이를 통해 QbD 적용 품목은 기존의 최종 제품 시험을 통한 품질관리 방식이 아니라, 위험평가에 기반해 과학적ㆍ통계적으로 설정된 공정별 품질관리 방식을 통해 실시간 출하시험 및 출하가 가능해진다.
아울러 임상ㆍ비임상시험 자료의 표준형식(CDISC) 제출 근거를 마련한다. 허가심사 시 임상시험과 비임상시험 기초자료 제출을 국제적으로 통용되는 국제표준양식인 CDISC에 따라 제출할 수 있게 된다.
또 신약 품목허가 신청 후 민원인이 허가ㆍ심사 담당 부서에 설명회의 등을 요청할 수 있는 ‘상담 신청제도’를 신설해 민원 처리의 투명성 및 예측성을 높인다.
수입 신약 제출자료 간소화도 추진된다. GMP 대상 수입 신약 허가 신청 시 제출해야 했던 수출국 정부 발행 ‘제조 및 판매증명서’ 제출을 면제해 해외 규제당국의 허가ㆍ해당 국가 내에서의 판매 여부와 별도로 평가할 수 있게 됐다.
식약처는 “이번 개정을 통해 제약업계의 국제경쟁력 강화를 유도하고 코로나19 등 공중보건 관련 신약의 신속 허가 및 공급을 지원할 수 있을 것으로 기대한다”라며 “앞으로도 안전성, 효과성 및 품질이 확보된 의약품을 신속하게 공급하기 위해 의약품 허가 편의성을 높이는 등 국민들이 안심하고 의약품을 사용할 수 있도록 관련 제도를 정비하겠다”라고 말했다.




![포켓몬, 아직도 '피카츄'만 아세요? [솔드아웃]](https://img.etoday.co.kr/crop/140/88/2296074.jpg)














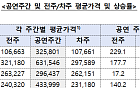
![삼성전자 지금 사도 될까…"설 이후 한 번 더 상승 여력" [찐코노미]](https://img.etoday.co.kr/crop/300/170/2296128.jpg)
![중국 춘절 연휴 시작, 북적이는 명동거리 [포토]](https://img.etoday.co.kr/crop/300/190/2296286.jpg)